Farbstoff-Aggregation
Durch die Absorption von Licht gelangen Farbstoffmoleküle in einen elektronisch angeregten Zustand. Dabei wird die aufgenommene Energie nur für eine kurze Zeit gespeichert und nach der Lebensdauer des Anregungszustands z.B. als Fluoreszenz wieder abgestrahlt.
In einer Farbstofflösung beeinflussen sich die als Punktdipole oder Oszillatoren anzusehenden angeregten Farbstoffmoleküle gegenseitig nicht, wenn die Abstände zwischen ihnen groß genug sind. Daher werden Absorption und Fluoreszenz der im Ensemble vorliegenden Chromophore nicht verändert.
Bei einem mittleren Abstand von ungefähr 5 – 10 nm zwischen den Chromophoren findet eine Beeinflussung nur über das „Strahlungsfeld“ der Oszillatoren, d.h. ohne direkten Kontakt, statt. Diese Art der Wechselwirkung zwischen zwei Farbstoffmolekülen wird z.B. durch das Modell der Förster-Resonanz-Energieübertragung (FRET) beschrieben.
Wenn der Abstand der Chromophore noch kleiner wird, z.B. in einer sehr konzentrierten Lösung, kann es zu einer starken gegenseitigen Beeinflussung durch die elektrostatischen Kräfte der Einzel-Oszillatoren kommen. Durch die intermolekulare Wechselwirkung der einzelnen Farbstoffmoleküle verändern sich sowohl Absorption- und Fluoreszenzverhalten einer solchen Farbstofflösung stark.
Rhodamin 6G in Wasser
Im UV/Vis-Spektrum einer konzentrierten wässrigen Lösung von Rhodamin 6G kann man an der kurzwelligen Flanke der Hauptabsorptionsbande das Auftreten einer Schulter beobachten. Ändert man die Konzentration (c) durch Verdünnen der Lösung und erhöht in gleicher Weise die Schichtdicke (d) der Küvette, so dass man nach dem Lambert-Beer-Gesetz immer die gleiche Extinktion erwarten könnte, stellt man folgenden Verlauf fest:
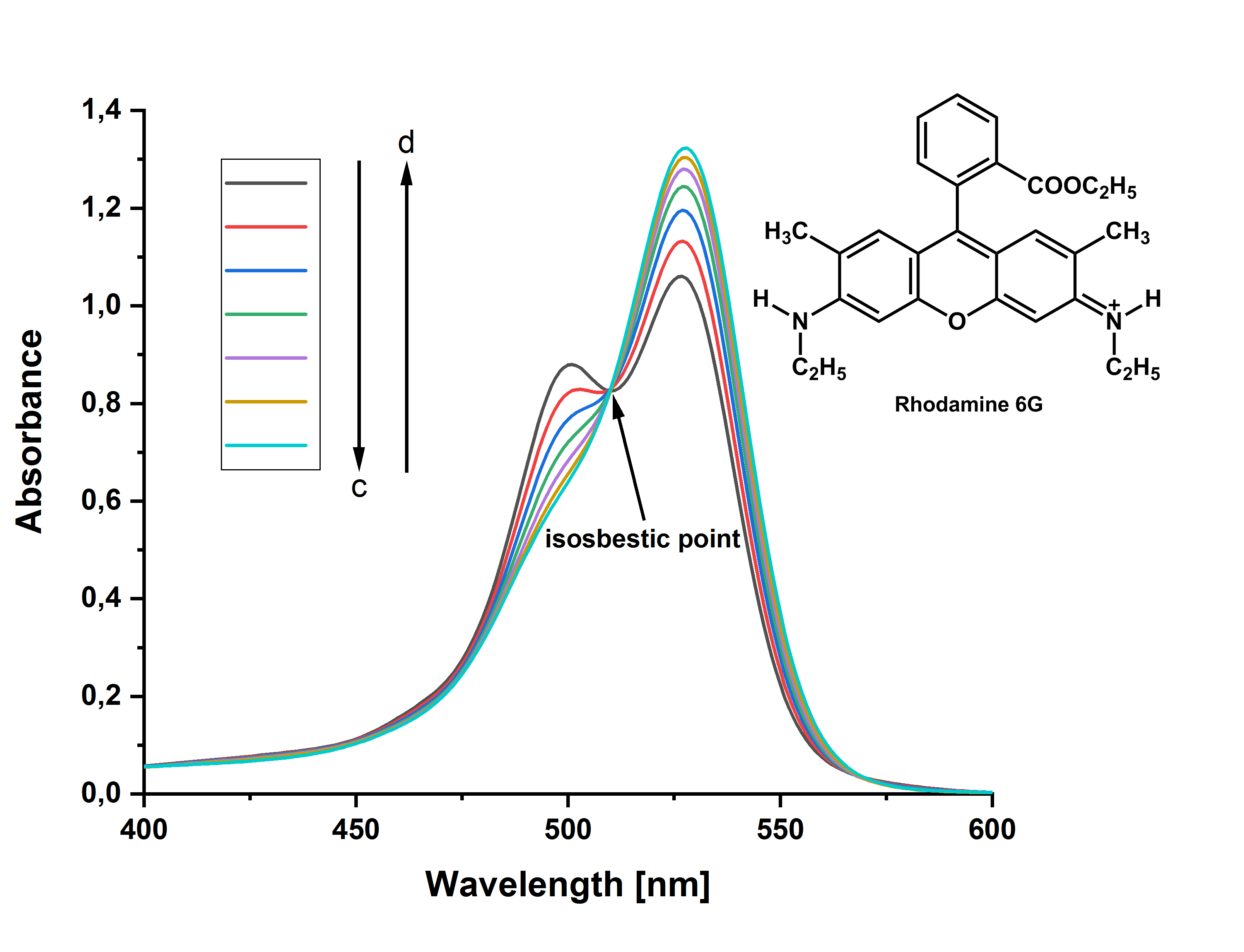
Das Auftreten eines isosbestischen Punktes – die Konzentrationsänderung aller beteiligten Stoffe ist linear, es gilt dE/dc = 0 – zeigt, dass sich zwei (oder mehr) Spezies definiert ineinander umwandeln bzw. miteinander im Gleichgewicht stehen. Es handelt sich also um ein dynamisches Gleichgewicht.

Die Dissoziations- oder Dimerisierungskonstante kann experimentell bestimmt werden: In einer Verdünnungsreihe, bei der die Verdünnung der Lösung immer durch die Änderung der Schichtdicke ausgeglichen wird, kann über die gemessene Extinktion am (Monomeren-)Maximum in Kenntnis des Verdünnungsfaktors und der Einwaagekonzentration jeweils der „effektive Extinktionskoeffizient“ ausgerechnet werden. Die Einwaagekonzentration bestimmt man aus dem UV-Spektrum einer stark verdünnten Lösung, bei der keine Dimerisation stattfindet. Da sich die Einzelabsorptionen im Lambert-Beer-Gesetz additiv verhalten, lässt sich mit der Reaktionsgleichung bzw. dem Massenwirkungsgesetz eine effektive Extinktion bzw. ein effektiver Extinktionskoeffizient formulieren. Durch parametrische Variation der Dissoziationskonstante und geeignete graphische Auftragung erhält man schließlich eine Gerade, aus deren Steigung und Achsenabschnitt die Extinktionskoeffizienten von Monomer und Dimer bestimmt werden können.
Hydrophobe Wechselwirkung
Die Aggregation von organischen Farbstoffen tritt besonders in Wasser oder Lösungsmitteln mit hoher Ionenstärke auf. Der Hauptgrund sind intermolekulare van der Waals-Kräfte: Durch die so genannte „hydrophobe Wechselwirkung“ versuchen die lipophilen Molekülen den hydrophilen Wassermolekülen „auszuweichen“, also der Hydrathülle eine möglichst geringe Oberfläche anzubieten. Dieses Phänomen ist auch für die Adsorption von z.B. Farbstoffen an Glasoberflächen oder unspezifische Bindungen an Substratmoleküle verantwortlich.
Die Tendenz zur Bildung von Dimeren oder höheren Aggregaten ist dabei abhängig von

Da es sich – wie oben beschrieben – um ein dynamisches Gleichgewicht handelt, lassen sich die Dimere durch Verdünnen der Lösung wieder in Monomere überführen. Das „Monomerenspektrum“ ist dann erreicht, wenn sich das gemessene Absorptionsspektrum bei weiterer Verdünnung und entsprechender Erhöhung der Schichtdicke nicht mehr ändert. Für die meisten hydrophoben ATTO-Farbstoffe ist dies bei einer Extinktion von ca. 0,04 (Schichtdicke 1 cm; c = 10-7 – 10-6 mol/l) gegeben.
Intramolekulare Wechselwirkung in Proteinkonjugaten / DOL-Bestimmung
Bei der Reaktion eines Farbstoff-NHS-Esters mit den Aminogruppen eines Proteins können Farbstoffkonjugate entstehen, in denen die kovalent gebundenen Farbstoffmoleküle eng benachbart sind und miteinander in Wechselwirkung treten können. Dies äußert sich in gleicher Weise durch eine starke Veränderung des Absorptionsspektrums, wie hier am Bespiel des ATTO 565-Steptavidinkonjugates deutlich zu erkennen ist:
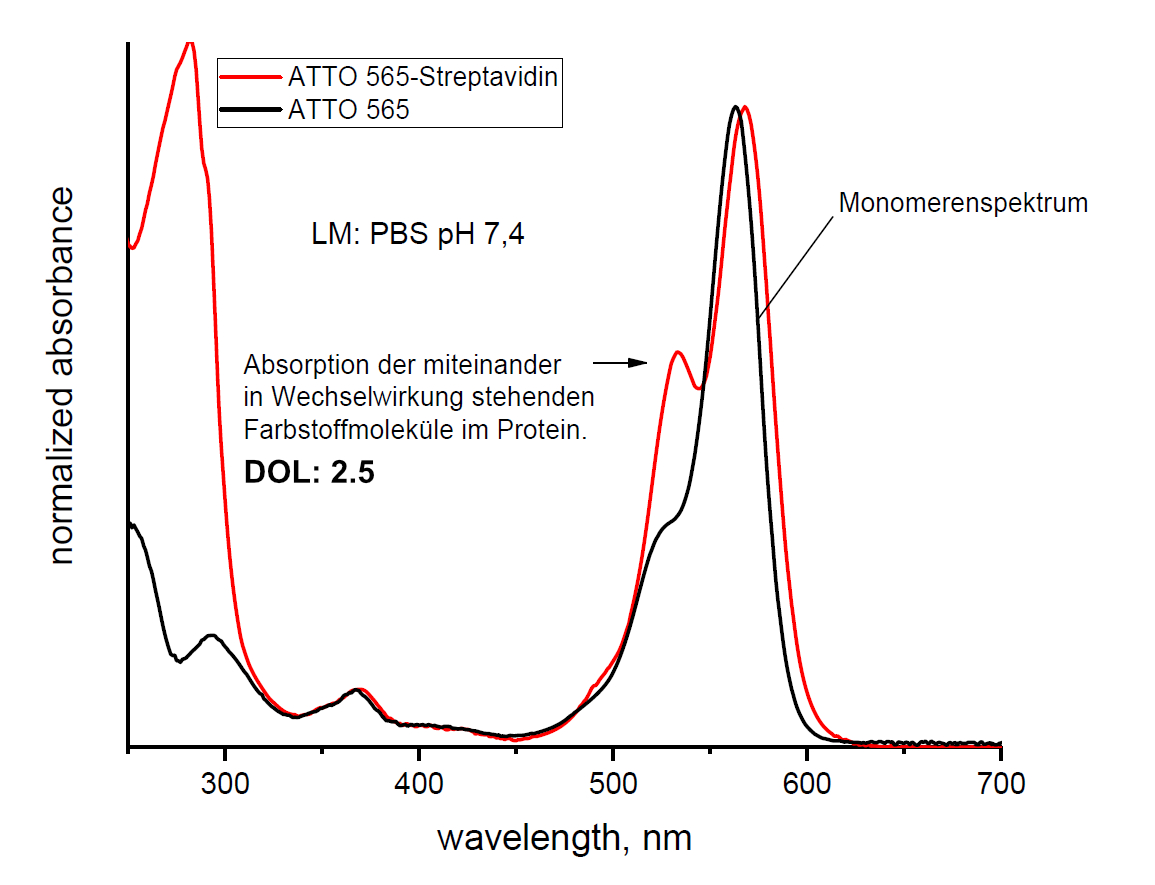
Man beobachtet im Konjugatspektrum eine zusätzliche, kurzwellige Absorptionsbande analog zur „Dimerenbande“ einer wässrigen Farbstofflösung mit hinreichend hoher Konzentration. Da es sich in diesem Fall um intramolekulare Wechselwirkungen der kovalent gebundenen Farbstoffmoleküle handelt, ändert sich das Absorptionsspektrum durch Verdünnen der Konjugatlösung nicht!
Die Bestimmung des Farbstoff-Protein-Verhältnisses (degree of labeling, DOL) für solche Fälle ist in unserer Arbeitsvorschrift „Proteinmarkierung“ beschrieben.
Man unterscheidet grundsätzlich zwischen zwei Formen von Aggregaten:
H-Aggregate (H = hypsochrom), kurzwellig
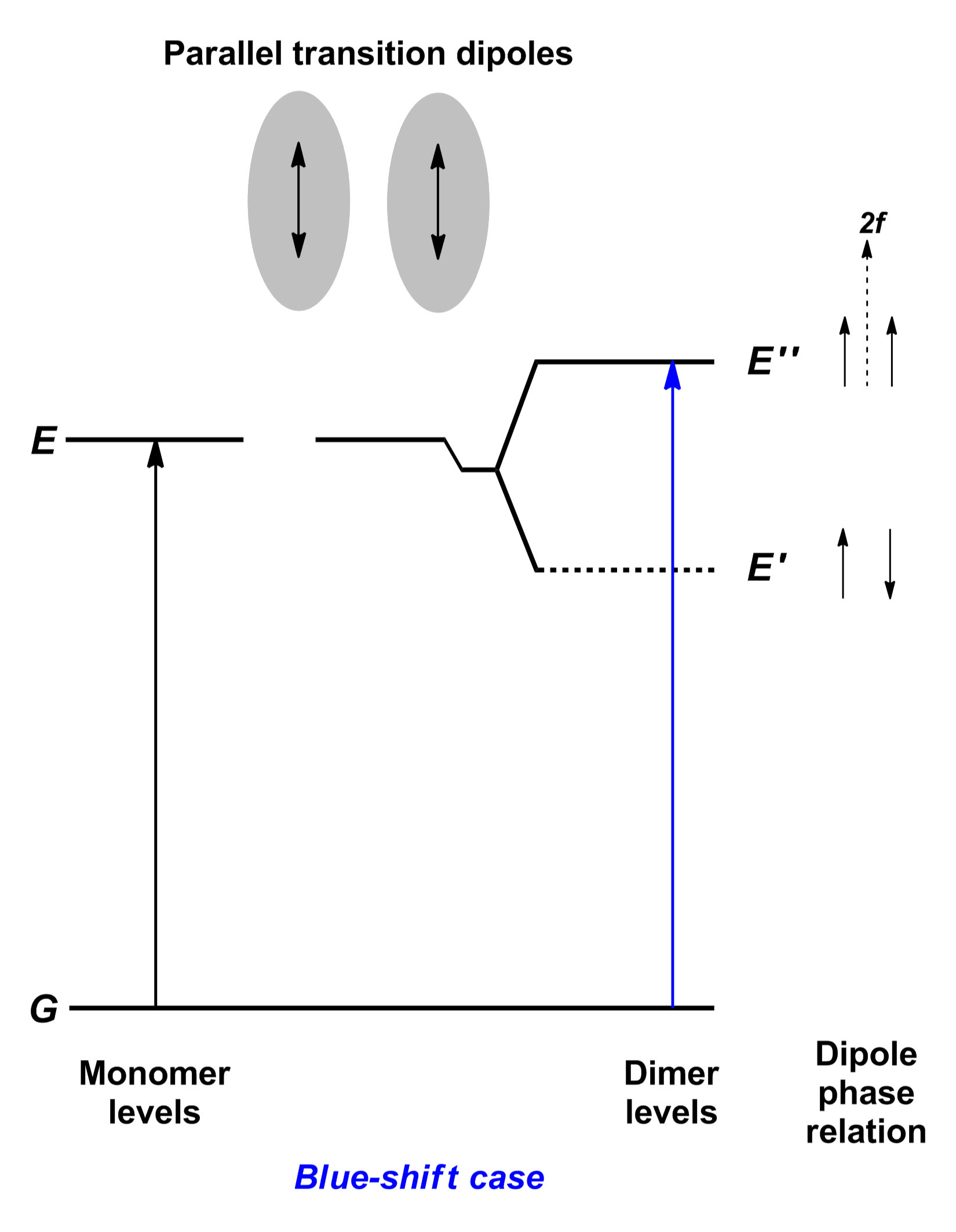
Diese Art der Aggregation tritt dann auf, wenn sich zwei oder mehr Farbstoffmoleküle so aneinander lagern, dass sich ihre Übergangsdipolmomente (beim S0 – S1 Übergang gewöhnlich entlang der Längsachse des chromophoren Systems verlaufend) parallel zu einander ausrichten. Man beobachtet eine – im Gegensatz zur Monomerenabsorption – hypsochrom verschobene Absorptionsbande.
Durch die räumliche Nähe beeinflussen sich die elektronischen Strukturen und man muss beide Moleküle quasi gemeinschaftlich betrachten. Es kommt zur Aufspaltung der Energieniveaus und der nun quantenmechanisch erlaubte Absorptionsübergang ist energiereicher und liegt damit bei kürzeren Wellenlängen. Aus diesem höheren Anregungszustand findet eine schnelle innere Umwandlung (IC, internal conversion) statt, so dass die Fluoreszenz gelöscht wird.
J-Aggregate (nach E.E. Jelley), langwellig
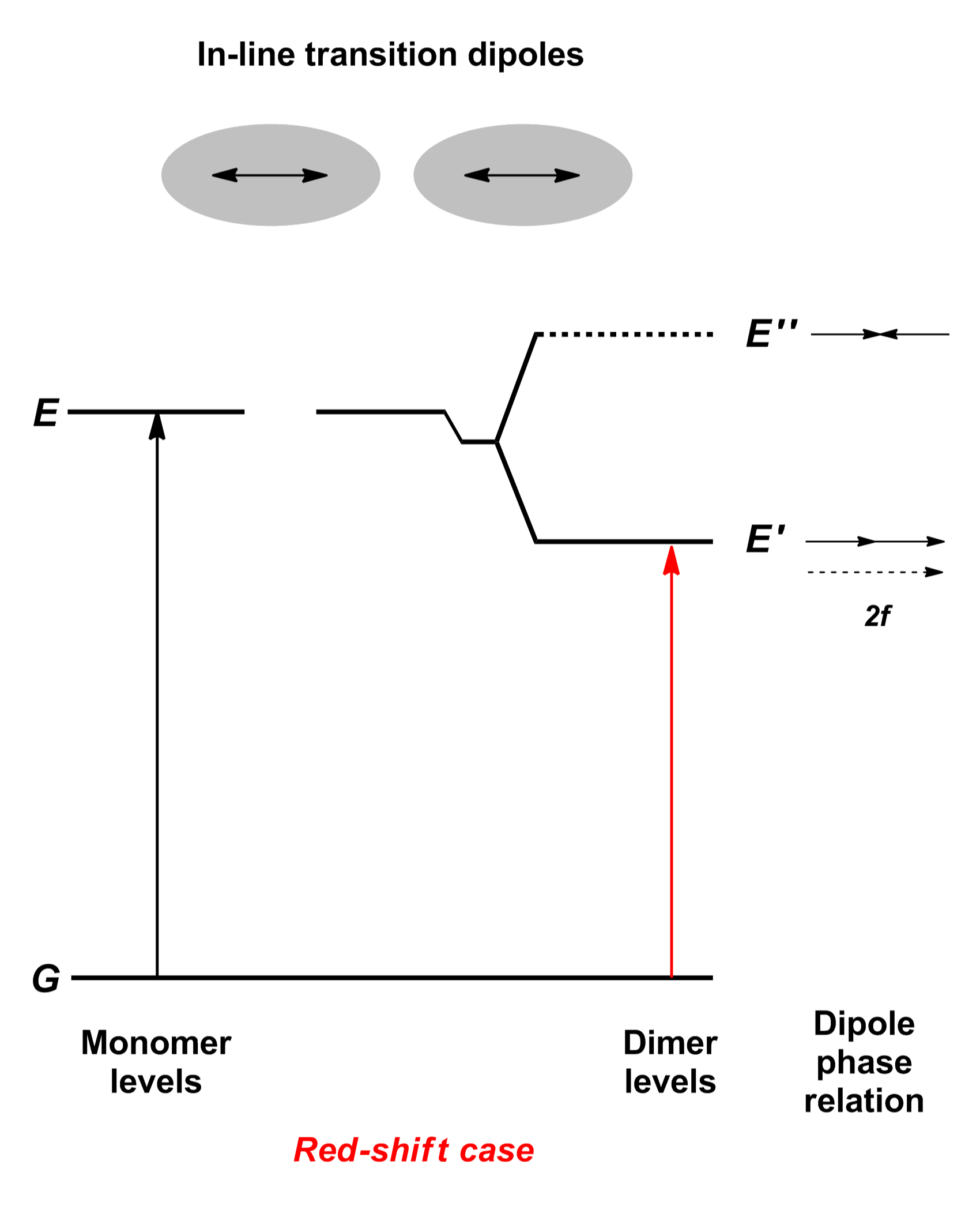
Bei dieser Art der Aggregation erhält man eine langwellige Verschiebung der Absorptionsbande, die mit einer deutlichen Verringerung der Halbwertsbreite der Bande verbunden ist.
J-Aggregate findet man häufig bei Polymethin-Farbstoffen, wie Cyaninen, Merocyaninen oder ähnlichen Chromophoren. Jelley und Scheibe beobachteten das Phänomen erstmals unabhängig voneinander am Farbstoff Pseudoisocyanin. Für die modellhafte Beschreibung des sich durch die Zusammenlagerung der Einzelfarbstoffe bildenden „supramolekularen Polymers“ wurden verschiedene Typen vorgeschlagen. Die einfachste Beschreibung der molekularen Verhältnisse stellt die Vorstellung dar, dass sich die einzelnen Moleküle quasi hintereinander anordnen, so dass auch die Übergangsdipolmomente auf einer Linie liegen. Aus der gemeinschaftlichen Betrachtung der Moleküle resultiert wieder eine Aufspaltung der Energieniveaus: Der quantenmechanisch erlaubte Übergang liegt jetzt energetisch niedriger, womit die langwellige Verschiebung der Absorptionsbande erklärt werden kann.
Die Aggregation lässt sich durch die Lösungsmittelzusammensetzung, den Zusatz von Salzen oder anderen Stoffen sowie die Konzentration stark beeinflussen. Unter idealen Bedingungen findet man im UV/Vis-Spektrum die beschriebene extrem schmale Absorptionsbande. Außerdem kann hier – im Gegensatz zu den H-Aggregaten – besonders bei tieferen Temperaturen durchaus Fluoreszenz beobachtet werden: Das Maximum der ebenfalls sehr schmalen Emissionsbande ist aber nur ein paar Nanometer langwelliger als das Absorptionsmaximum.
In der Literatur ist – je nach den experimentellen Bedingungen – auch die „Verbreiterung“ der Absorptionsbande beschrieben, die u.a. durch die Einbeziehung des verbotenen Elektronenübergangs des J-Aggregats erklärt wird.
ATTO 488 markierte Phospholipide
Lösungen von ATTO 488 markierten Phospholipiden in reinem Chloroform überraschen zunächst durch ihre unerwartete Farbe: Statt hellgelb mit leuchtend grüner Fluoreszenz erscheint die stumpfe Lösung pink bis magenta. Die langwellig verschobene Absorption kann mit dem Vorliegen von J-Aggregaten erklärt werden. Beim Verdünnen der betreffenden Lösung mit Methanol verändert sich die Farbe zum gewohnten Gelbton und die starke Fluoreszenz wird sichtbar. Durch die Veränderung der Lösungsmittelzusammensetzung werden die Aggregate zurückgedrängt.
Die folgenden Abbildungen zeigen Lösungen von ATTO 488 markiertem 1,2-Dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) in reinem Chloroform und in einem Lösungsmittelgemisch aus Chloroform/Methanol (8:2, V/V):


Links sind beide Lösungen im normalen Tageslicht dargestellt. Rechts ist die grüne Fluoreszenz der mit Methanol versetzten Lösung beim Bestrahlen mit UV-Licht (366 nm) besonders gut zu erkennen.
Literatur:
E. Jelley, Spectral Absorption and Fluorescence of Dyes in the Molecular State, Nature 138, 1009 (1936).
G. Scheibe, Über die Veränderlichkeit der Absorptionsspektren in Lösungen und die Nebenvalenzen als ihre Ursache, Angewandte Chemie 50, 212 (1937).
T. Förster, Energiewanderung und Fluoreszenz, Die Naturwissenschaften 33, 166 (1946).
M. Kasha, H.R. Rawls, M. Ashraf El-Bayoumi, The Exciton Model in Molecular Spectroscopy, Pure Appl. Chem. 11, 371 (1965).
O. Valdes-Aguilera, D.C. Neckers, Aggregation Phenomena in Xanthene Dyes, Acc. Chem. Res. 22, 171 (1989).
J. Hernando et al., Excitonic Behavior of Rhodamine Dimers: A Single-Molecule Study, J. Phys. Chem. A 107, 43 (2003).
V.I. Gavrilenko, M.A. Noginov, Ab initio study of optical properties of rhodamine 6G molecular dimers, J. Chem. Phys. 124, 0044301 (2006).
In einer Farbstofflösung beeinflussen sich die als Punktdipole oder Oszillatoren anzusehenden angeregten Farbstoffmoleküle gegenseitig nicht, wenn die Abstände zwischen ihnen groß genug sind. Daher werden Absorption und Fluoreszenz der im Ensemble vorliegenden Chromophore nicht verändert.
Bei einem mittleren Abstand von ungefähr 5 – 10 nm zwischen den Chromophoren findet eine Beeinflussung nur über das „Strahlungsfeld“ der Oszillatoren, d.h. ohne direkten Kontakt, statt. Diese Art der Wechselwirkung zwischen zwei Farbstoffmolekülen wird z.B. durch das Modell der Förster-Resonanz-Energieübertragung (FRET) beschrieben.
Wenn der Abstand der Chromophore noch kleiner wird, z.B. in einer sehr konzentrierten Lösung, kann es zu einer starken gegenseitigen Beeinflussung durch die elektrostatischen Kräfte der Einzel-Oszillatoren kommen. Durch die intermolekulare Wechselwirkung der einzelnen Farbstoffmoleküle verändern sich sowohl Absorption- und Fluoreszenzverhalten einer solchen Farbstofflösung stark.
Rhodamin 6G in Wasser
Im UV/Vis-Spektrum einer konzentrierten wässrigen Lösung von Rhodamin 6G kann man an der kurzwelligen Flanke der Hauptabsorptionsbande das Auftreten einer Schulter beobachten. Ändert man die Konzentration (c) durch Verdünnen der Lösung und erhöht in gleicher Weise die Schichtdicke (d) der Küvette, so dass man nach dem Lambert-Beer-Gesetz immer die gleiche Extinktion erwarten könnte, stellt man folgenden Verlauf fest:
Das Auftreten eines isosbestischen Punktes – die Konzentrationsänderung aller beteiligten Stoffe ist linear, es gilt dE/dc = 0 – zeigt, dass sich zwei (oder mehr) Spezies definiert ineinander umwandeln bzw. miteinander im Gleichgewicht stehen. Es handelt sich also um ein dynamisches Gleichgewicht.
Die Dissoziations- oder Dimerisierungskonstante kann experimentell bestimmt werden: In einer Verdünnungsreihe, bei der die Verdünnung der Lösung immer durch die Änderung der Schichtdicke ausgeglichen wird, kann über die gemessene Extinktion am (Monomeren-)Maximum in Kenntnis des Verdünnungsfaktors und der Einwaagekonzentration jeweils der „effektive Extinktionskoeffizient“ ausgerechnet werden. Die Einwaagekonzentration bestimmt man aus dem UV-Spektrum einer stark verdünnten Lösung, bei der keine Dimerisation stattfindet. Da sich die Einzelabsorptionen im Lambert-Beer-Gesetz additiv verhalten, lässt sich mit der Reaktionsgleichung bzw. dem Massenwirkungsgesetz eine effektive Extinktion bzw. ein effektiver Extinktionskoeffizient formulieren. Durch parametrische Variation der Dissoziationskonstante und geeignete graphische Auftragung erhält man schließlich eine Gerade, aus deren Steigung und Achsenabschnitt die Extinktionskoeffizienten von Monomer und Dimer bestimmt werden können.
Hydrophobe Wechselwirkung
Die Aggregation von organischen Farbstoffen tritt besonders in Wasser oder Lösungsmitteln mit hoher Ionenstärke auf. Der Hauptgrund sind intermolekulare van der Waals-Kräfte: Durch die so genannte „hydrophobe Wechselwirkung“ versuchen die lipophilen Molekülen den hydrophilen Wassermolekülen „auszuweichen“, also der Hydrathülle eine möglichst geringe Oberfläche anzubieten. Dieses Phänomen ist auch für die Adsorption von z.B. Farbstoffen an Glasoberflächen oder unspezifische Bindungen an Substratmoleküle verantwortlich.
Die Tendenz zur Bildung von Dimeren oder höheren Aggregaten ist dabei abhängig von
- der Konzentration des Farbstoffs – je höher die Konzentration, desto stärker die Aggregation
- dem Lösungsmittel – in Wasser oder Methanol kann im Gegensatz zu Ethanol oder anderen organischen Lösungsmitteln häufig Aggregation beobachtet werden. Dies zeigt eindrucksvoll die Gegenüberstellung der Absorptionsspektren gleich konzentrierter Lösungen von ATTO 565 in wässrigem PBS-Puffer (pH 7,4) und Ethanol mit Trifluoressigsäure (TFAc):
- eventuell vorhandenen Elektrolyten (Salzen), vor allem beim Auftreten von Ionenpaaren in organischen Lösungsmitteln wie Chloroform
- der Temperatur – bei höherer Temperatur wird durch die thermische Bewegung die Aggregation erschwert
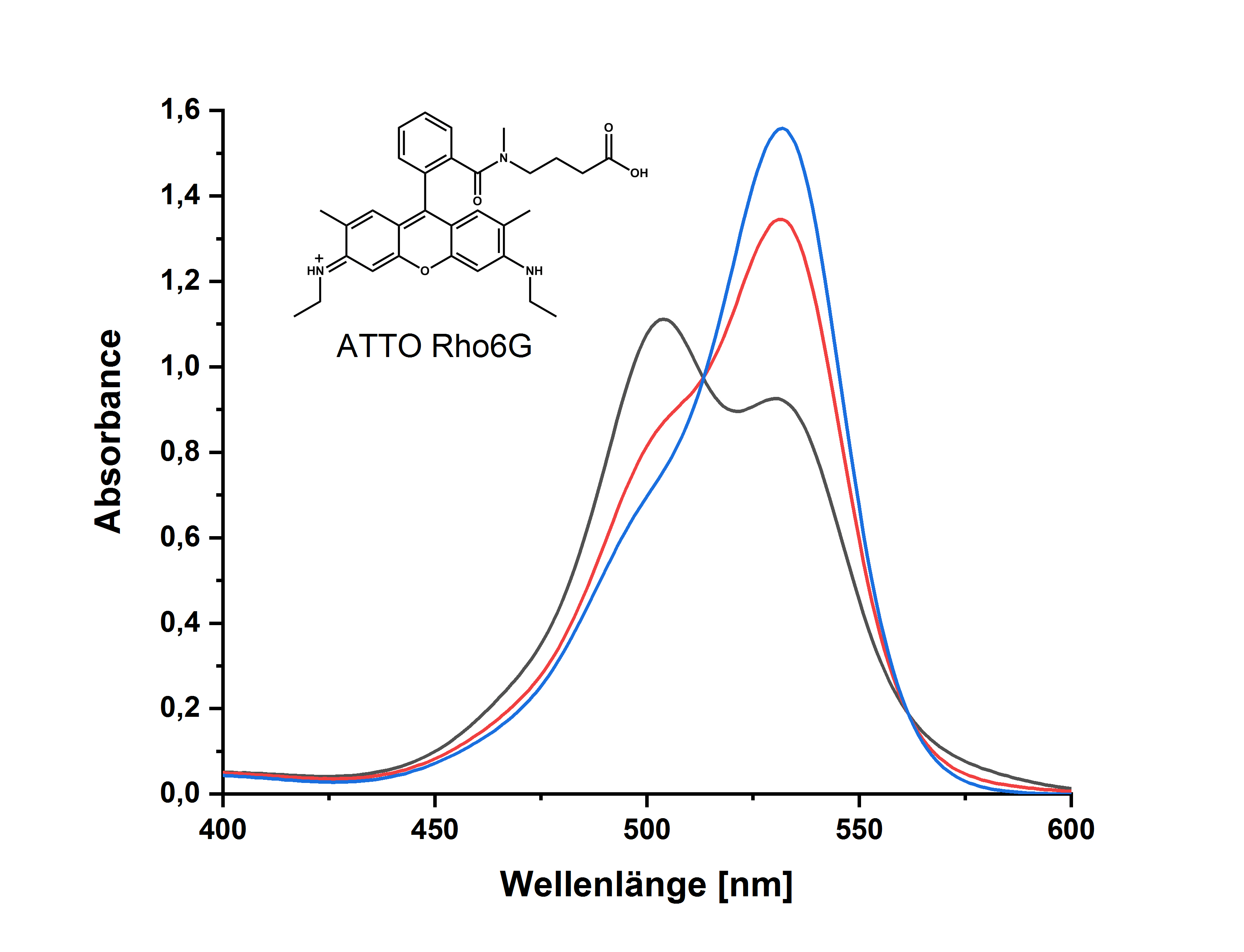
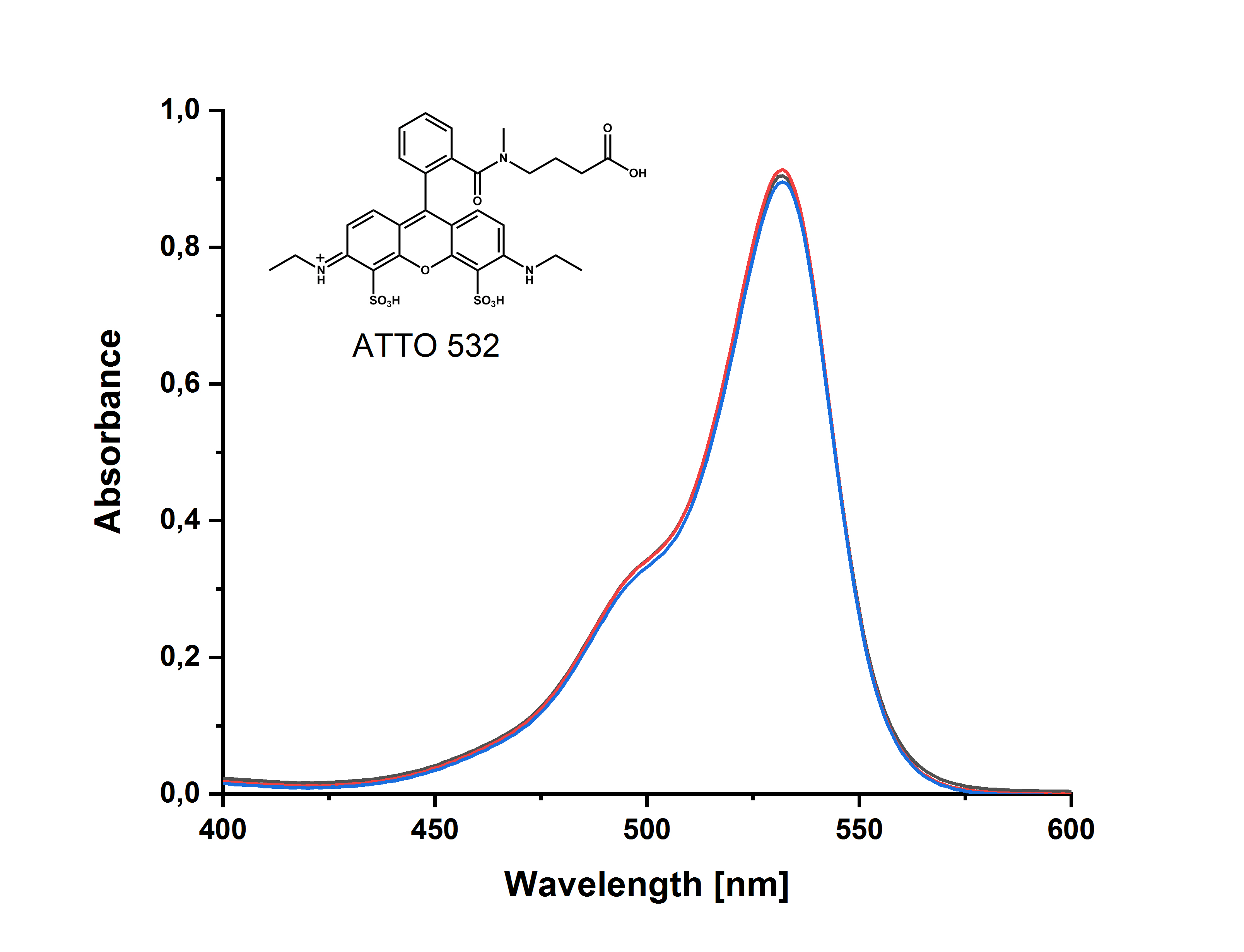
- der molekularen Struktur des Farbstoffs – Farbstoffe mit hydrophilen Gruppen, wie z.B. ATTO 488, ATTO 532, ATTO 542 etc., zeigen im Gegensatz zu hydrophoberen Farbstoffen wie ATTO Rho6G, ATTO Rho11, ATTO Rho12 etc. in wässriger Lösung keine Aggregation:
Da es sich – wie oben beschrieben – um ein dynamisches Gleichgewicht handelt, lassen sich die Dimere durch Verdünnen der Lösung wieder in Monomere überführen. Das „Monomerenspektrum“ ist dann erreicht, wenn sich das gemessene Absorptionsspektrum bei weiterer Verdünnung und entsprechender Erhöhung der Schichtdicke nicht mehr ändert. Für die meisten hydrophoben ATTO-Farbstoffe ist dies bei einer Extinktion von ca. 0,04 (Schichtdicke 1 cm; c = 10-7 – 10-6 mol/l) gegeben.
Intramolekulare Wechselwirkung in Proteinkonjugaten / DOL-Bestimmung
Bei der Reaktion eines Farbstoff-NHS-Esters mit den Aminogruppen eines Proteins können Farbstoffkonjugate entstehen, in denen die kovalent gebundenen Farbstoffmoleküle eng benachbart sind und miteinander in Wechselwirkung treten können. Dies äußert sich in gleicher Weise durch eine starke Veränderung des Absorptionsspektrums, wie hier am Bespiel des ATTO 565-Steptavidinkonjugates deutlich zu erkennen ist:
Man beobachtet im Konjugatspektrum eine zusätzliche, kurzwellige Absorptionsbande analog zur „Dimerenbande“ einer wässrigen Farbstofflösung mit hinreichend hoher Konzentration. Da es sich in diesem Fall um intramolekulare Wechselwirkungen der kovalent gebundenen Farbstoffmoleküle handelt, ändert sich das Absorptionsspektrum durch Verdünnen der Konjugatlösung nicht!
Die Bestimmung des Farbstoff-Protein-Verhältnisses (degree of labeling, DOL) für solche Fälle ist in unserer Arbeitsvorschrift „Proteinmarkierung“ beschrieben.
Man unterscheidet grundsätzlich zwischen zwei Formen von Aggregaten:
H-Aggregate (H = hypsochrom), kurzwellig
Diese Art der Aggregation tritt dann auf, wenn sich zwei oder mehr Farbstoffmoleküle so aneinander lagern, dass sich ihre Übergangsdipolmomente (beim S0 – S1 Übergang gewöhnlich entlang der Längsachse des chromophoren Systems verlaufend) parallel zu einander ausrichten. Man beobachtet eine – im Gegensatz zur Monomerenabsorption – hypsochrom verschobene Absorptionsbande.
Durch die räumliche Nähe beeinflussen sich die elektronischen Strukturen und man muss beide Moleküle quasi gemeinschaftlich betrachten. Es kommt zur Aufspaltung der Energieniveaus und der nun quantenmechanisch erlaubte Absorptionsübergang ist energiereicher und liegt damit bei kürzeren Wellenlängen. Aus diesem höheren Anregungszustand findet eine schnelle innere Umwandlung (IC, internal conversion) statt, so dass die Fluoreszenz gelöscht wird.
J-Aggregate (nach E.E. Jelley), langwellig
Bei dieser Art der Aggregation erhält man eine langwellige Verschiebung der Absorptionsbande, die mit einer deutlichen Verringerung der Halbwertsbreite der Bande verbunden ist.
J-Aggregate findet man häufig bei Polymethin-Farbstoffen, wie Cyaninen, Merocyaninen oder ähnlichen Chromophoren. Jelley und Scheibe beobachteten das Phänomen erstmals unabhängig voneinander am Farbstoff Pseudoisocyanin. Für die modellhafte Beschreibung des sich durch die Zusammenlagerung der Einzelfarbstoffe bildenden „supramolekularen Polymers“ wurden verschiedene Typen vorgeschlagen. Die einfachste Beschreibung der molekularen Verhältnisse stellt die Vorstellung dar, dass sich die einzelnen Moleküle quasi hintereinander anordnen, so dass auch die Übergangsdipolmomente auf einer Linie liegen. Aus der gemeinschaftlichen Betrachtung der Moleküle resultiert wieder eine Aufspaltung der Energieniveaus: Der quantenmechanisch erlaubte Übergang liegt jetzt energetisch niedriger, womit die langwellige Verschiebung der Absorptionsbande erklärt werden kann.
Die Aggregation lässt sich durch die Lösungsmittelzusammensetzung, den Zusatz von Salzen oder anderen Stoffen sowie die Konzentration stark beeinflussen. Unter idealen Bedingungen findet man im UV/Vis-Spektrum die beschriebene extrem schmale Absorptionsbande. Außerdem kann hier – im Gegensatz zu den H-Aggregaten – besonders bei tieferen Temperaturen durchaus Fluoreszenz beobachtet werden: Das Maximum der ebenfalls sehr schmalen Emissionsbande ist aber nur ein paar Nanometer langwelliger als das Absorptionsmaximum.
In der Literatur ist – je nach den experimentellen Bedingungen – auch die „Verbreiterung“ der Absorptionsbande beschrieben, die u.a. durch die Einbeziehung des verbotenen Elektronenübergangs des J-Aggregats erklärt wird.
ATTO 488 markierte Phospholipide
Lösungen von ATTO 488 markierten Phospholipiden in reinem Chloroform überraschen zunächst durch ihre unerwartete Farbe: Statt hellgelb mit leuchtend grüner Fluoreszenz erscheint die stumpfe Lösung pink bis magenta. Die langwellig verschobene Absorption kann mit dem Vorliegen von J-Aggregaten erklärt werden. Beim Verdünnen der betreffenden Lösung mit Methanol verändert sich die Farbe zum gewohnten Gelbton und die starke Fluoreszenz wird sichtbar. Durch die Veränderung der Lösungsmittelzusammensetzung werden die Aggregate zurückgedrängt.
Die folgenden Abbildungen zeigen Lösungen von ATTO 488 markiertem 1,2-Dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) in reinem Chloroform und in einem Lösungsmittelgemisch aus Chloroform/Methanol (8:2, V/V):
Links sind beide Lösungen im normalen Tageslicht dargestellt. Rechts ist die grüne Fluoreszenz der mit Methanol versetzten Lösung beim Bestrahlen mit UV-Licht (366 nm) besonders gut zu erkennen.
Literatur:
E. Jelley, Spectral Absorption and Fluorescence of Dyes in the Molecular State, Nature 138, 1009 (1936).
G. Scheibe, Über die Veränderlichkeit der Absorptionsspektren in Lösungen und die Nebenvalenzen als ihre Ursache, Angewandte Chemie 50, 212 (1937).
T. Förster, Energiewanderung und Fluoreszenz, Die Naturwissenschaften 33, 166 (1946).
M. Kasha, H.R. Rawls, M. Ashraf El-Bayoumi, The Exciton Model in Molecular Spectroscopy, Pure Appl. Chem. 11, 371 (1965).
O. Valdes-Aguilera, D.C. Neckers, Aggregation Phenomena in Xanthene Dyes, Acc. Chem. Res. 22, 171 (1989).
J. Hernando et al., Excitonic Behavior of Rhodamine Dimers: A Single-Molecule Study, J. Phys. Chem. A 107, 43 (2003).
V.I. Gavrilenko, M.A. Noginov, Ab initio study of optical properties of rhodamine 6G molecular dimers, J. Chem. Phys. 124, 0044301 (2006).