Absorption/Fluoreszenz
Neben Brechungs-, Streuungs-, Interferenz- und Beugungsphänomenen an teilweise oder vollständig transparenter Materie, die einen Farbeindruck erzeugen können, wie z.B. beim Regenbogen, dem Himmelsblau, schimmernden Seifenblasen, prächtigen Pfauenfedern oder dünnen Ölfilmen und Schichten, entstehen die Farben in unserer Umwelt vorrangig durch die Absorption bzw. Reflexion von Licht.
„Farbigkeit“ von Materie entsteht dabei durch selektive Absorption von Licht aus dem sichtbaren Teil des Spektrums der elektromagnetischen Strahlung. Dieser Bereich liegt zwischen ultraviolettem und infrarotem Licht. Auf molekularer Ebene bedeutet dies, dass Moleküle in einen elektronisch angeregten Zustand gelangen: Durch die Absorption eines Lichtquants (Photon) wird ein Elektron aus dem höchsten besetzten Molekülorbital (HOMO = highest occupied molecular orbital; Energie E1) in das niedrigste unbesetzte Molekülorbital (LUMO = lowest unoccupied molecular orbital; Energie E2) angehoben.
EPhoton = h ⋅ υ = (h ⋅c) / λ = ΔE = E2 - E1
EPhoton: Energie des Photons
h: Planck-Konstante
υ: Frequenz der Strahlung
c: Lichtgeschwindigkeit
λ: Wellenlänge der Strahlung
ΔE: Energieunterschied der beiden Niveaus
UV/Vis-Spektroskopie
Die Absorption von Licht – z.B. in einer Farbstofflösung – kann gemessen werden. Grundlage für die quantitative UV/Vis-Spektroskopie ist das Lambert-Beer-Gesetz:
E = lg(I0 / I) = ε ⋅ c ⋅ d
E: Extinktion, optische Dichte (OD) oder englisch Absorbance (ABS)
ε: molarer Extinktionskoeffizient
c: Konzentration
d: Schichtdicke
I0: Lichtintensität vor der Probe
I: Lichtintensität nach der Probe
c = E / (ε ⋅ d)
Das Lambert-Beer-Gesetzt gilt für monochromatische Strahlung. Durch die Auftragung der Extinktion über der Wellenlänge erhält man für verschiedene Wellenlängen das UV/Vis-Spektrum einer Verbindung.
Das Lambert-Beer-Gesetz verliert seine Gültigkeit bei höher konzentrierten Lösungen sowie beim Auftreten von konzentrationsabhängigen Reaktionen, wie es z.B. die Aggregation von Farbstoffen darstellt.
Lizenz: CC BY-SA 4.0
Marmot2019, https://commons.wikimedia.org/wiki/File:Transmittance.svg?
Beim Durchgang durch die Lösung wird der Lichtstrahl abgeschwächt, ein Teil des Lichts ist von Molekülen in der Lösung absorbiert worden, d.h. es gilt bei Vernachlässigung von Streuung und Reflexion:
A + T = 1 = (I0 – I)/I0 + I/I0
A = Absorption = absorbierter Teil des Lichts = (I0 – I) / I0
T = Transmission = durchgelassener Teil des Lichts = I / I0
Daher gilt:
E = -lg (T)
Die Proportionalitätskonstante im Lambert-Beer-Gesetz ist eine wellenlängenabhängige Stoffkonstante und beschreibt die so genannte Übergangswahrscheinlichkeit mit der eine Anregung erlaubt ist. Es gelten spektroskopische Auswahlregeln, d.h. man unterscheidet zwischen erlaubten und verbotenen Übergängen. Als wichtigste Auswahlregel gilt die Erhaltung der Multiplizität (siehe unten). Dabei haben die erlaubten Übergänge eine größere Übergangswahrscheinlichkeit und damit einen höheren Extinktionskoeffizienten. Bei organischen Farbstoffen liegt der Extinktionskoeffizient am langwelligen Absorptionsmaximum bei Werten > 105 L mol-1 cm-1; es handelt sich um erlaubte Übergänge innerhalb des konjugierten π-Systems.
Die Absorption ist ein sehr schneller Prozess (10-15 bis 10-14 s), der ohne Spinumkehr verläuft. Die durch die Absorption des Photons vom Molekül aufgenommene Energie wird nach einer beschränkten Lebensdauer des Anregungszustandes in unterschiedlicher Form wieder abgegeben. Schließt man photochemische Reaktionen aus, so ist neben der Abgabe in Form von thermischer Energie auch das Auftreten einer Lumineszenzerscheinung als Fluoreszenz oder Phosphoreszenz möglich. Diese photophysikalischen Elementarprozesse, bei denen die chemische Identität des Moleküls am Ende erhalten bleibt, lassen sich graphisch in Form eines Jablonski-Diagramms oder Jablonski-Termschemas darstellen.
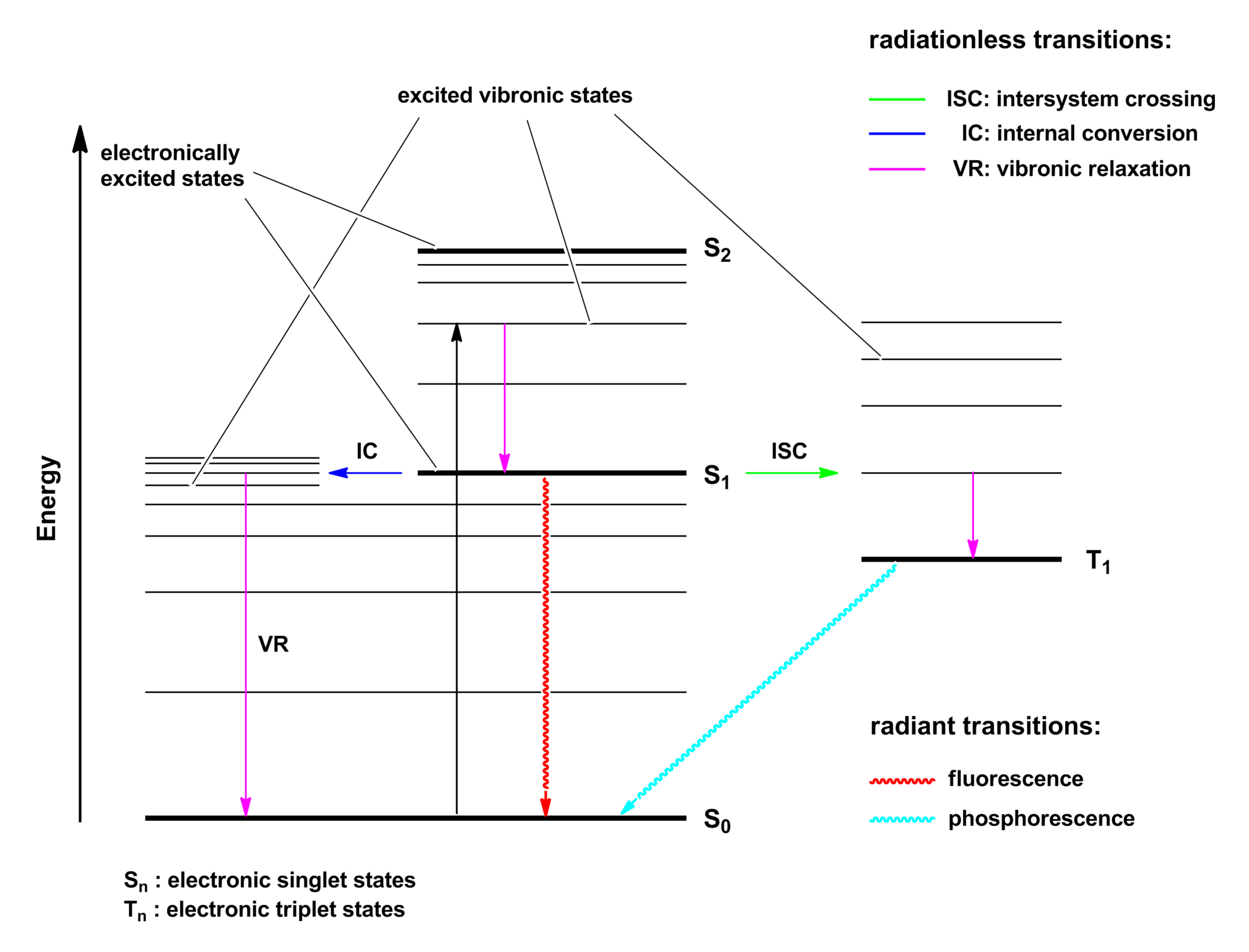
Darin sind neben den Elektronenniveaus S0 – S2 und T1 auch die zugehörigen Schwingungsunterniveaus dargestellt. Man unterscheidet bei der Darstellung zwischen Strahlungsprozessen (Wellenlinien) und strahlungslosen Prozessen (Striche).
Ein Singulettzustand S besitzt nur gepaarte Elektronen, der Gesamtspin ist S = 0; ein Triplettzustand T besitzt zwei ungepaarte Elektronen, der Gesamtspin ist S = 1; daraus ergibt sich die Multiplizität M = 2 S + 1 (Anzahl der Energieniveaus in Anwesenheit eines Magnetfelds). Für Singulettzustände ist M = 1 und für Triplettzustände ist M = 3. Die Auswahlregel der Erhaltung der Multiplizität erlaubt somit nur Übergänge innerhalb des Singulett- oder Triplettsystems („Spinverbot“).
Fluoreszenz
Fluoreszenz (10-9 bis 10-7 s) stellt die Lichtemission dar, die bei einer Desaktivierung des angeregten Moleküls durch Übergang vom niedrigsten Schwingungsniveau des ersten angeregten Singulettzustandes S1 zu Schwingungsniveaus des elektronischen Grundzustandes S0 (d.h. zwischen Zuständen gleicher Multiplizität) beobachtbar ist („Regel von Kasha“). Somit ist die beobachtete Fluoreszenz unabhängig von der Anregungswellenlänge.
Das Auftreten von Fluoreszenz, die von G.G. Stokes nach dem lumineszierenden Mineral Fluorit (CaF2) so bezeichnet wurde, lässt sich empirisch mit bestimmten Strukturmerkmalen der organischen Moleküle in Beziehung setzen: nichtaromatische Kohlenwasserstoffe zeigen keine Fluoreszenz; selbst dann nicht, wenn sie wie im Falle des Lycopins elf konjugierte Doppelbindungen aufweisen, während aromatische Verbindungen fast ausnahmslos fluoreszenzfähig sind.
Eine bei „Aromaten“ häufig gegebene starre und ebene Molekülstruktur darf daher in vielen - wenn auch nicht allen - Fällen als Voraussetzung für eine gute Fluoreszenzfähigkeit angesehen werden.
Weiterhin hat sich gezeigt, dass die Einführung gewisser Gruppen in aromatische Kohlenwasserstoffe die Fluoreszenz schwächt. Dazu zählt u.a. die Nitro-Gruppe, welche bewirkt, dass z.B. Nitrobenzol nicht fluoresziert.
Interkombination (ISC)
Aber auch Brom- oder Jodatome verringern die Fluoreszenz. Man spricht dann vom inneren Schweratomeffekt. Diese Substituenten erleichtern durch eine größere Spin-Bahn-Kopplung den eigentlich spinverbotenen Übergang in das Triplett-System. Dieser wechselseitige Übergang zwischen isoenergetischen Schwingungsniveaus des Singulett-Systems und des Triplett-Systems wird als „intersystem crossing“ (ISC) bezeichnet. Paramagnetische Substanzen mit ungepaarten Elektronen, wie beispielsweise molekularer Sauerstoff O2 vermitteln als Katalysatoren ebenfalls diesen Übergang.
Phosporeszenz
Eine bei der Desaktivierung des Schwingungsgrundzustandes des Triplett-Zustandes T1 in Schwingungsniveaus des elektronischen Grundzustandes S0 auftretende Lichtemission bezeichnet man als Phosphoreszenz. Im Unterschied zur Fluoreszenz, die nur solange beobachtbar ist, wie eine Anregung erfolgt, kann Phosphoreszenz u.U. auch noch längere Zeit nach Beendigung der Anregung auftreten bzw. andauern („Nachleuchten“). Dies hängt wiederum mit dem Spinverbot zusammen, was eine längere Lebensdauer des Triplett-Zustandes (10-4 bis 102 s) verursacht und damit eine verzögerte Desaktivierung in den Singulett-Grundzustand zur Folge hat.
Stokes-Verschiebung
Da Fluoreszenz oder Phosphoreszenz immer aus dem Schwingungsgrundzustand v0 des elektronisch angeregten Zustands S1 oder T1 erfolgen, ist die Emissionsbande gegenüber der Anregungsbande immer zu höheren Wellenlängen, d.h. bathochrom, verschoben. Die Phosphoreszenz ist im Vergleich zur Fluoreszenz dabei noch stärker langwellig verschoben.
Internal Conversion (IC)
Weiterhin kann eine „internal conversion“ (IC, Innere Umwandlung) genannte Desaktivierung z.B. durch einen Übergang vom Schwingungsgrundzustand des ersten elektronisch angeregten Zustandes S1 in isoenergetische vibronische Niveaus des elektronischen Grundzustandes S0 erfolgen. Aus solchen höher angeregten Vibrationsniveaus erfolgt dann eine Schwingungsrelaxation (10-12 s) unter Abgabe von Wärme. Diese Art der strahlungslosen Deaktivierung ist stets schneller als Lumineszenzvorgänge. Besonders effektiv scheint dieser Prozess bei sehr beweglichen Molekülen zu sein. Deshalb findet man bei aliphatischen Verbindungen fast ausschließlich die innere Umwandlung bis zum elektronischen Grundzustand und keine Fluoreszenz. Auch bei den meisten anderen Molekülen dominieren strahlungslose Desaktivierungsprozesse gegenüber Lumineszenzvorgängen.
Fluoreszenzfarbstoffe
Nur in sehr wenigen organischen Farbstoffen ist die strahlungslose Desaktivierung langsam genug, so dass Übergänge vom angeregten Zustand in den Grundzustand an Bedeutung gewinnen, bei denen die überschüssige Energie durch Emission eines Photons als Fluoreszenz abgegeben wird.
Ein Fluoreszenzfarbstoff ist durch seine spektroskopischen Eigenschaften wie Anregungs- und Emissionsspektrum, Fluoreszenzquantenausbeute (ηfl) und Fluoreszenzabklingzeit (τfl) charakterisiert. Die Fluoreszenz des Farbstoffs ist unabhängig von der Wellenlänge der Anregung.
Was bei der Auswahl eines Farbstoffs als Fluoreszenzlabel zu beachten ist, haben wir in der entsprechenden Rubrik dargestellt.
Spektrale Eigenschaften
Die spektralen Eigenschaften eines Fluoreszenzfarbstoffes sind sehr stark abhängig von der Molekülstruktur. Damit die Absorption eines Moleküls im sichtbaren Wellenlängenbereich (400 - 700 nm) liegt, muss die Energiedifferenz zwischen Grund- und angeregtem Zustand hinreichend niedrig sein. Das auffälligste Strukturmerkmal eines organischen Farbstoffs ist ein konjugiertes π-Elektronensystem, der so genannte „Chromophor“ (griech. Farbträger).
In den meisten Fällen ist das Fluoreszenzspektrum eines Farbstoffes in erster Näherung ein Spiegelbild der längstwelligen Absorptionsbande, wobei die Fluoreszenz typischerweise um 25 - 40 nm zu längeren Wellen verschoben ist.
Absorption von Rhodamin-Farbstoffen
Bei den meisten ATTO-Farbstoffen besitzt der Chromophor ein starres molekulares Grundgerüst. Viele Vertreter gehören zur Familie der Rhodamin-Farbstoffe. Diese Farbstoffe besitzen als gemeinsames Strukturelement einen Carboxyphenyl-Substituenten am zentralen Kohlenstoffatom eines Xanthen-Grundkörpers.
Die Carboxyl-Gruppe (rot) befindet sich in der 2- oder ortho-Position und beeinflusst maßgeblich die physikalisch-chemischen Eigenschaften aller Rhodamine.
Durch die freie ortho-Carboxyl-Gruppe haben z.B. ATTO 565 und ATTO 590 sowie deren Derivate besondere Eigenschaften, die beim Umgang mit diesen beiden Farbstoffen zu beachten sind. Für die Verwendung als Fluoreszenzmarker besitzen ATTO 565 und ATTO 590 noch eine zusätzliche Carboxyl-Gruppe in der 4- oder 5-Position des Phenyl-Substituenten:
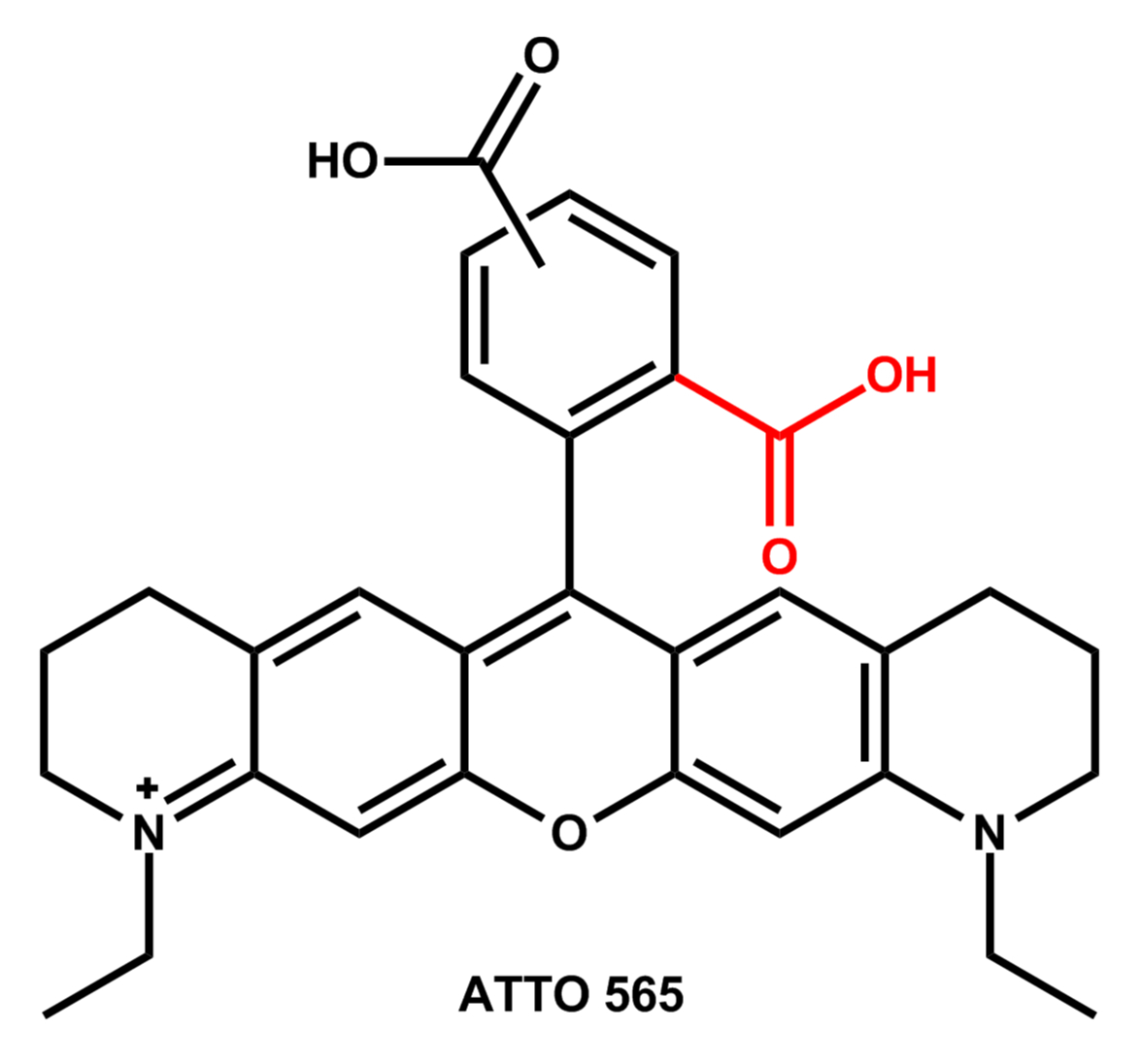
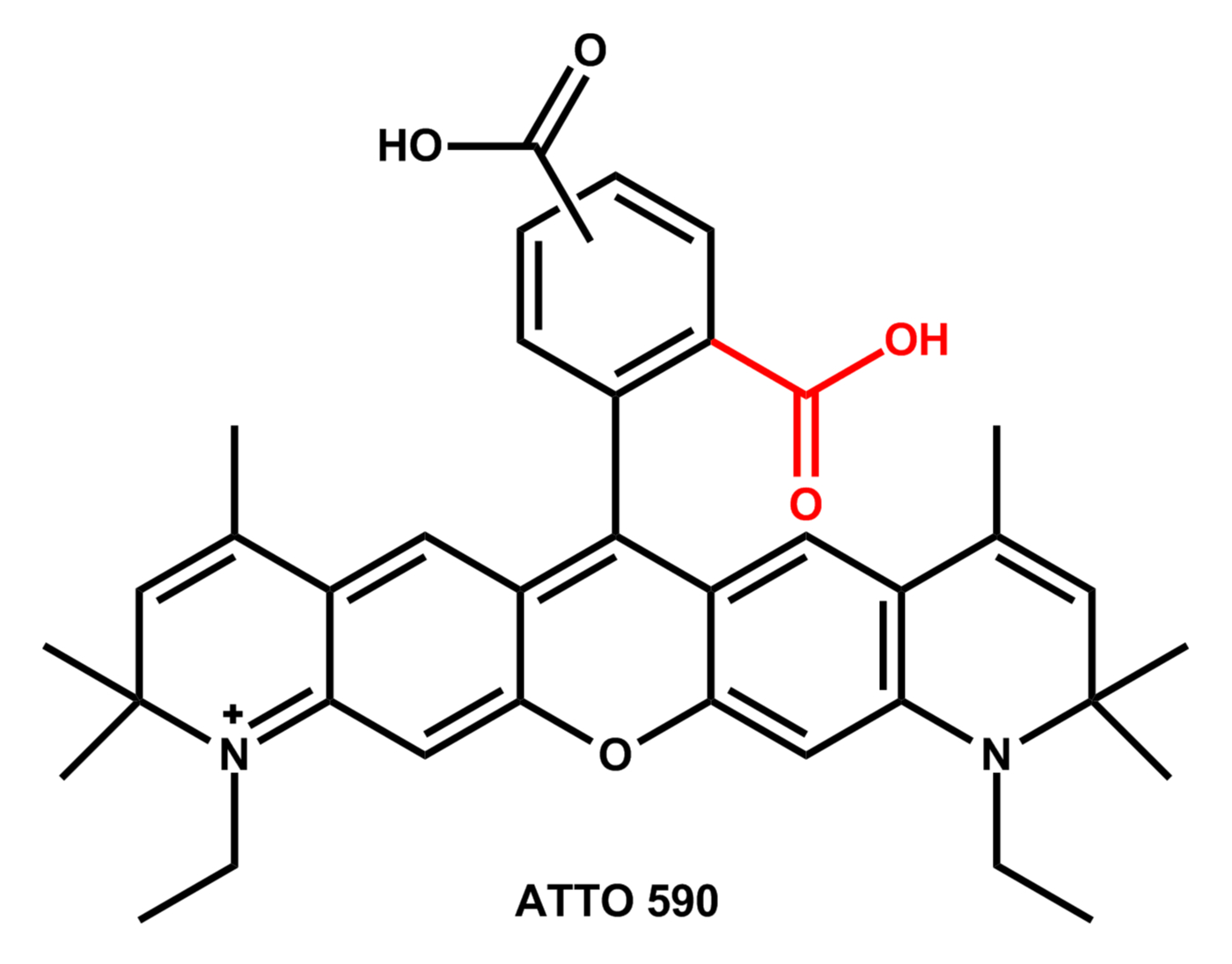
Abhängigkeit der Absorptionswellenlänge vom pH-Wert
Das Protonierungs-Deprotonierungs-Gleichgewicht der ortho-Carboxyl-Gruppe beeinflusst die optischen Eigenschaften der Rhodamine durch die Nähe zum Chromophor sehr stark.
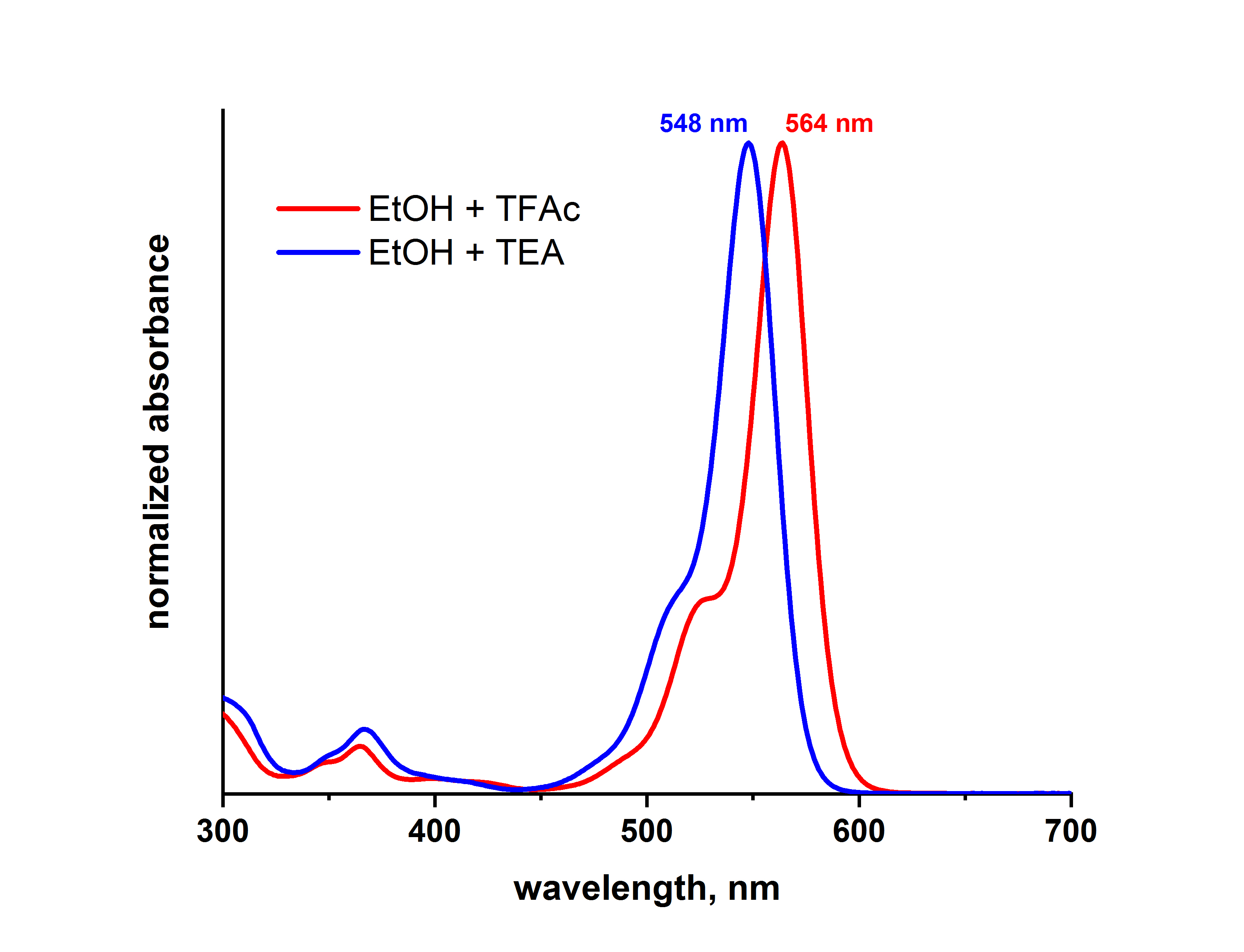
So ist die Lage des Absorptionsmaximums der Farbstoffe ATTO 565 und ATTO 590 für die protonierte und deprotonierte Farbstoffform verschieden. Beispielsweise verschiebt sich das Absorptionsmaximum von ATTO 565 in Ethanol durch Deprotonierung (Zugabe von Triethylamin) beider Carboxylgruppen relativ zur protonierten Form (Zugabe von Trifluoressigsäure) um 16 nm zu kürzeren Wellen (hypsochrom):
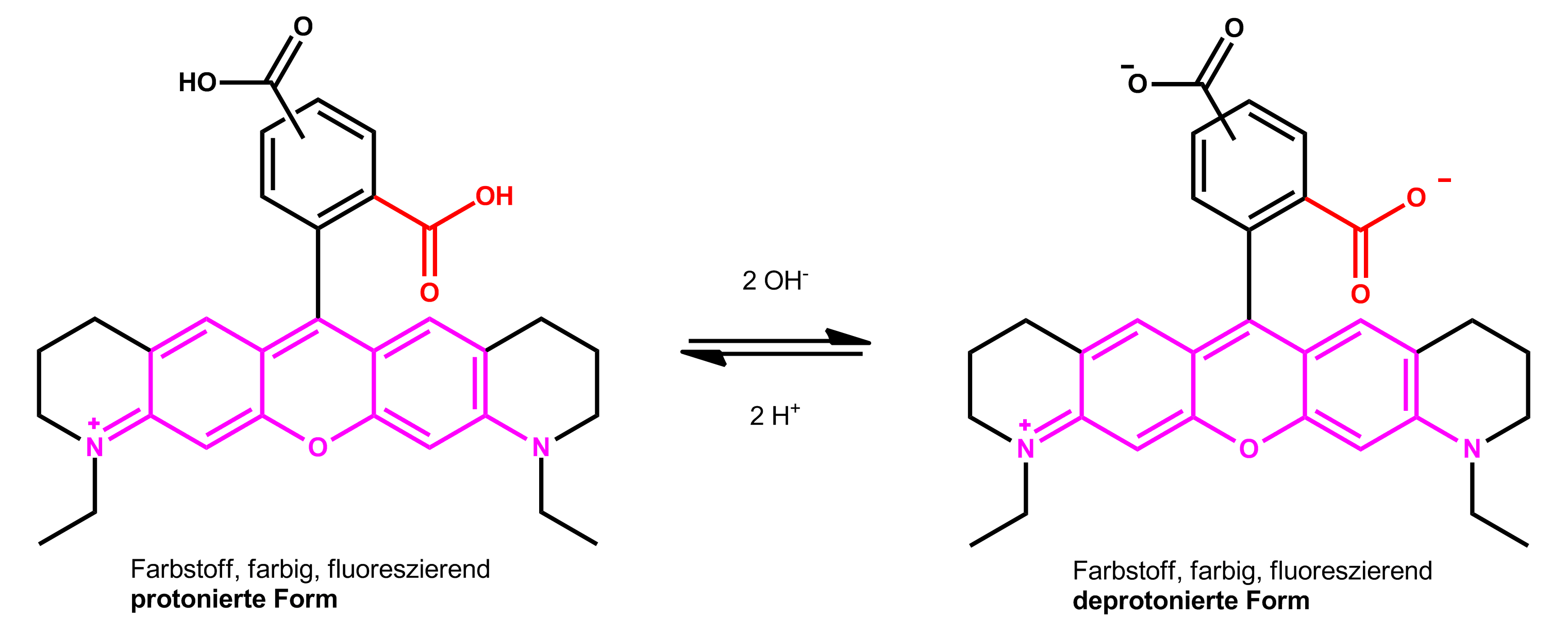
Farbstoff-Spirolacton-Gleichgewicht
Im Gleichgewicht können ATTO 565 und ATTO 590 in der deprotonierten Form aber auch ein farbloses Spirolacton bilden: Beim nukleophilen Angriff des Carboxylat-Anions am zentralen Kohlenstoff entsteht ein Fünfring und das chromophore System des Farbstoffs wird unterbrochen, so dass die sich bildende Verbindung im Sichtbaren nicht mehr absorbiert und fluoresziert:
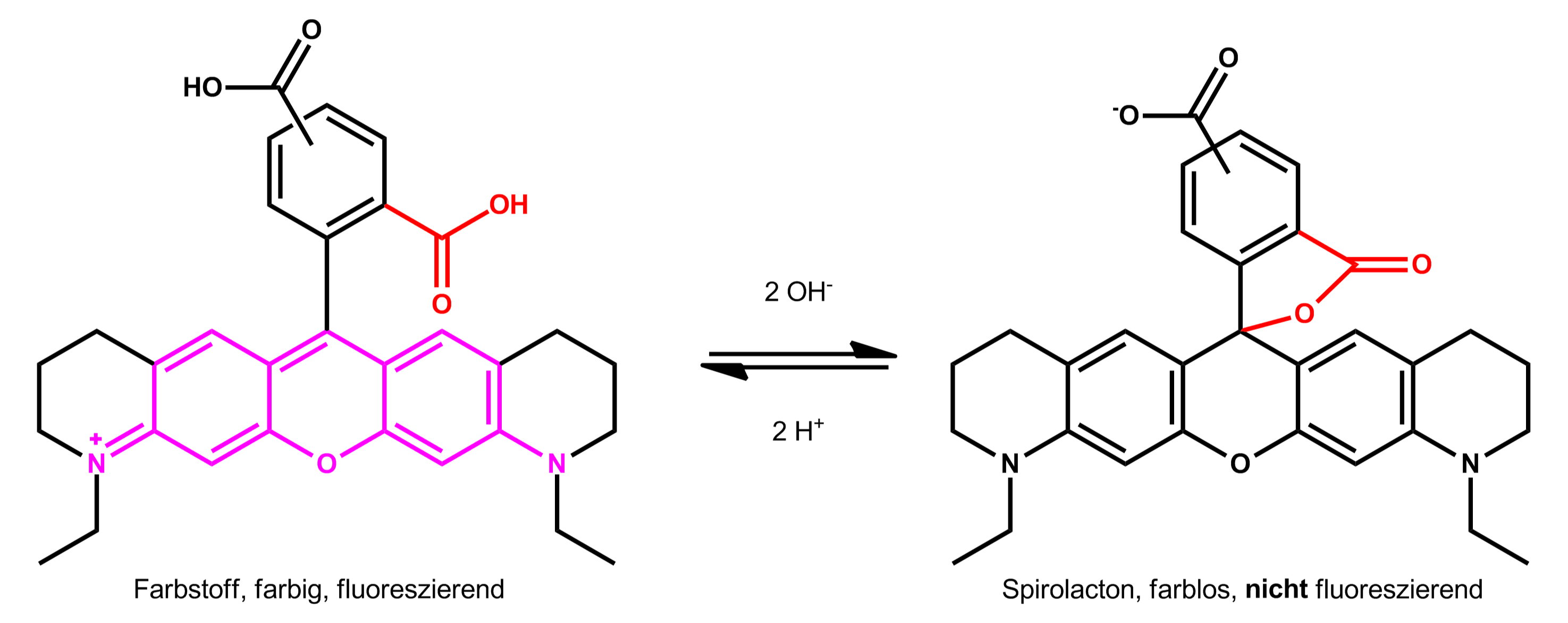
Der Anteil an Spirolacton bzw. die Lage dieses Gleichgewichts ist stark abhängig vom Lösungsmittel, vom pH-Wert, von der Temperatur und der Farbstoffstruktur. Das Gleichgewicht liegt in polaren, aprotischen Lösungsmitteln fast vollständig auf der Seite des Spirolactons; in wasserfreiem Aceton sind Lösungen der Farbstoffe ATTO 565 und ATTO 590 deshalb nahezu farblos. In polaren, protischen Lösungsmitteln wie Wasser oder Ethanol liegt das Gleichgewicht – wie oben beschrieben – hingegen fast vollständig auf der Seite der farbigen Form.
Das Fluoreszenzspektrum
In den meisten Fällen ist das Fluoreszenzspektrum eines Farbstoffes ein Spiegelbild der langwelligen Absorptionsbande, wie hier am Beispiel von ATTO 514 sehr schön zu sehen ist:
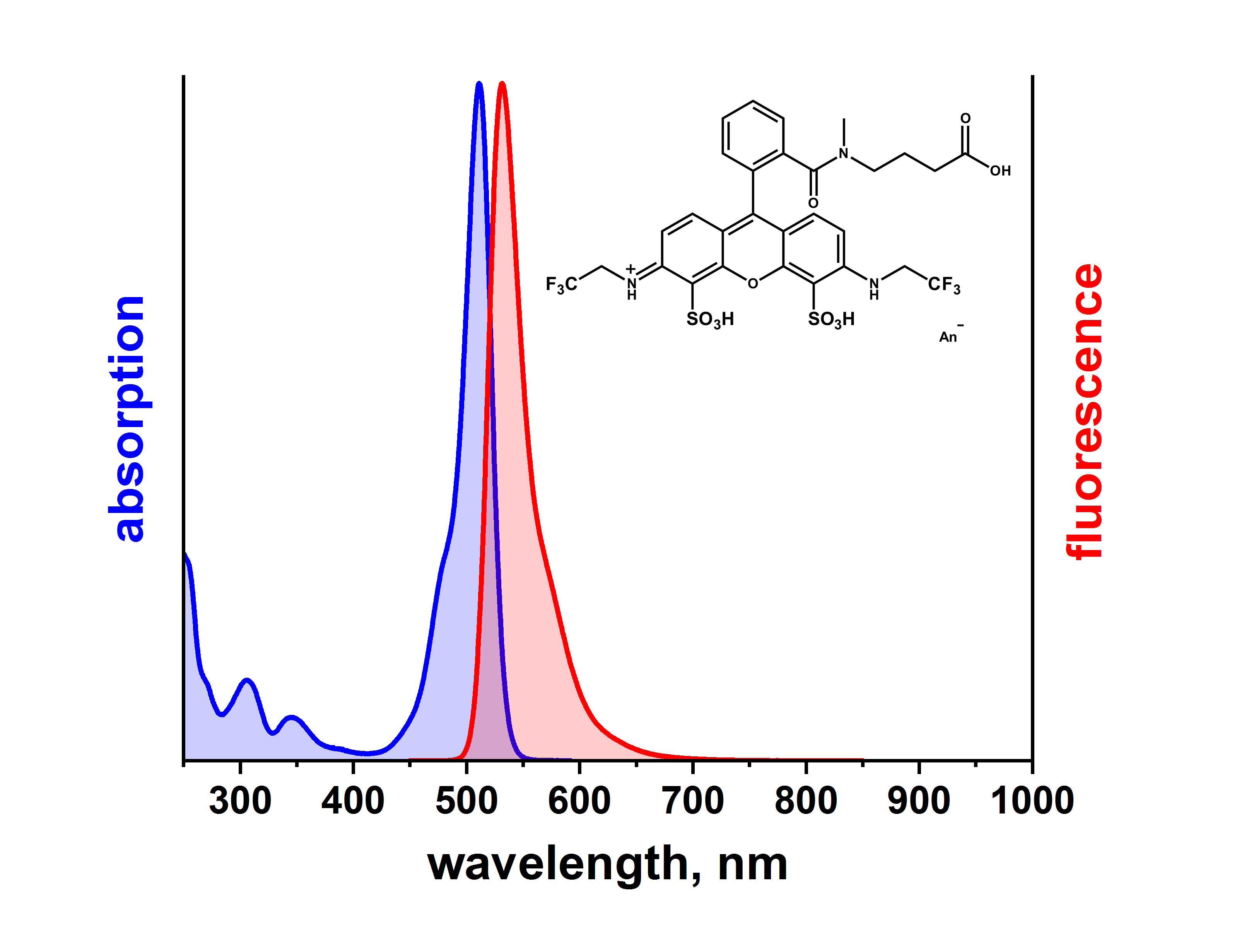
Das Fluoreszenzmaximum ist dabei gegenüber dem Absorptionsmaximum typischerweise um 25 - 40 nm zu längeren Wellen verschoben. Dieser so genannte Stokes-Shift wird außerdem noch durch die Umorientierung der den Farbstoff umgebenden Lösungsmittelmoleküle während der Lebensdauer des angeregten Zustands beeinflusst, so dass die Emission aus einem energieärmeren „Lösungsmittel-relaxiertem Zustand“ erfolgt. Je stärker sich die Elektronenverteilung während der Anregung ändert, desto größer wird dieser Energieunterschied und damit auch der Stokes-Shift. Dies erfährt bei unseren ATTO LS-Farbstoffen eine praktische Anwendung.
Fluoreszenzquantenausbeute (ηfl)
Eine der wichtigsten Eigenschaften eines Fluoreszenzfarbstoffes ist die Fluoreszenzquantenausbeute (ηfl). Die Quantenausbeute beschreibt das Verhältnis von emittierten Photonen (nfl) zur Zahl der absorbierten Photonen (nabs).
ηfl = nfl / nabs
Somit kann die Fluoreszenzquantenausbeute 100 % nie überschreiten. Wie oben angedeutet stehen strahlungslose Desaktivierungsprozesse sehr häufig in Konkurrenz zur Fluoreszenz und verringern damit die Quantenausbeute.
Für die Untersuchung eines Farbstoffs mittels Fluoreszenz ist natürlich eine hohe Fluoreszenzquantenausbeute von Vorteil und wünschenswert.
Auf die experimentelle Bestimmung der Fluoreszenzquantenausbeute gehen wir an anderer Stelle genauer ein.
Fluoreszenzabklingzeit (τfl)
Die Emission eines Fluoreszenzphotons nach der Anregung ist ein statistischer Vorgang. Somit ist die Dauer, für die ein einzelnes Molekül im angeregten Zustand verbleibt, ebenfalls eine statistische Größe. Betrachtet man jedoch ein Ensemble von vielen identischen Molekülen, so ergibt sich eine wohldefinierte Zerfallsstatistik. Nach erfolgter Anregung mittels eines kurzen Laserpulses nimmt die Zahl der Moleküle im einfachsten Fall exponentiell ab. Die Zeit, nach der die Zahl der angeregten Moleküle (n1) auf den Faktor 1/e (ca. 37%) abgesunken ist, nennt man Fluoreszenzabklingzeit (τfl).
n1(t) = n1(0) exp(- t / τfl)
Die Fluoreszenzabklingzeit ist eine wichtige Eigenschaft eines Farbstoffs und kann zu seiner Identifizierung verwendet werden. Werte von τfl liegen bei unseren ATTO-Farbstoffen typischerweise im Bereich von Nanosekunden.
Die Bestimmung der Fluoreszenzabklingzeit ist an anderer Stelle beschrieben.
Molekulare Wechselwirkungen
Die Fluoreszenzabklingzeit, wie auch die Fluoreszenzquantenausbeute eines Farbstoffes, ist keine unveränderliche Größe, sondern wird durch die Umgebung des Farbstoffes, d.h. Lösungsmittel und Temperatur, beeinflusst. Abklingzeit und Quantenausbeute des Farbstoffes sind dabei nicht unabhängig voneinander, sondern sind über folgende Beziehung miteinander verknüpft:
τfl = τ0 × ηfl
τ0 ist die sogenannte natürliche Abklingzeit, die im Fall fehlender strahlungsloser Desaktivierung (ηfl = 100%) auftreten würde.
Demzufolge können Änderungen in der Fluoreszenzabklingzeit Auskünfte über Änderungen der lokalen Umgebung des Farbstoffmoleküls liefern.
Allgemeine Literatur:
M.F. Vitha, Spectroscopy, Principles and Instrumentation, Wiley-VCH, 2018, ISBN 978-1-119-43664-5.
T. Förster, Fluoreszenz organischer Verbindungen, Vandenhoeck & Ruprecht, Göttingen, 1997 (Reprint), ISBN 3525423128
K. H. Drexhage, Structure and Properties of Laser Dyes, in: F. P. Schäfer, Dye Lasers, Springer Verlag, Berlin, Heidelberg, 1973.
M. Klessinger, J. Michl, Excited States and Photochemistry of Organic Molecules, Wiley-VCH, Weinheim, 1995, ISBN 0471185765.
W.A. Bingel, Theorie der Molekülspektren, Verlag Chemie, Weinheim, 1967.
Textbooks of Physical and Theoretical Chemistry.